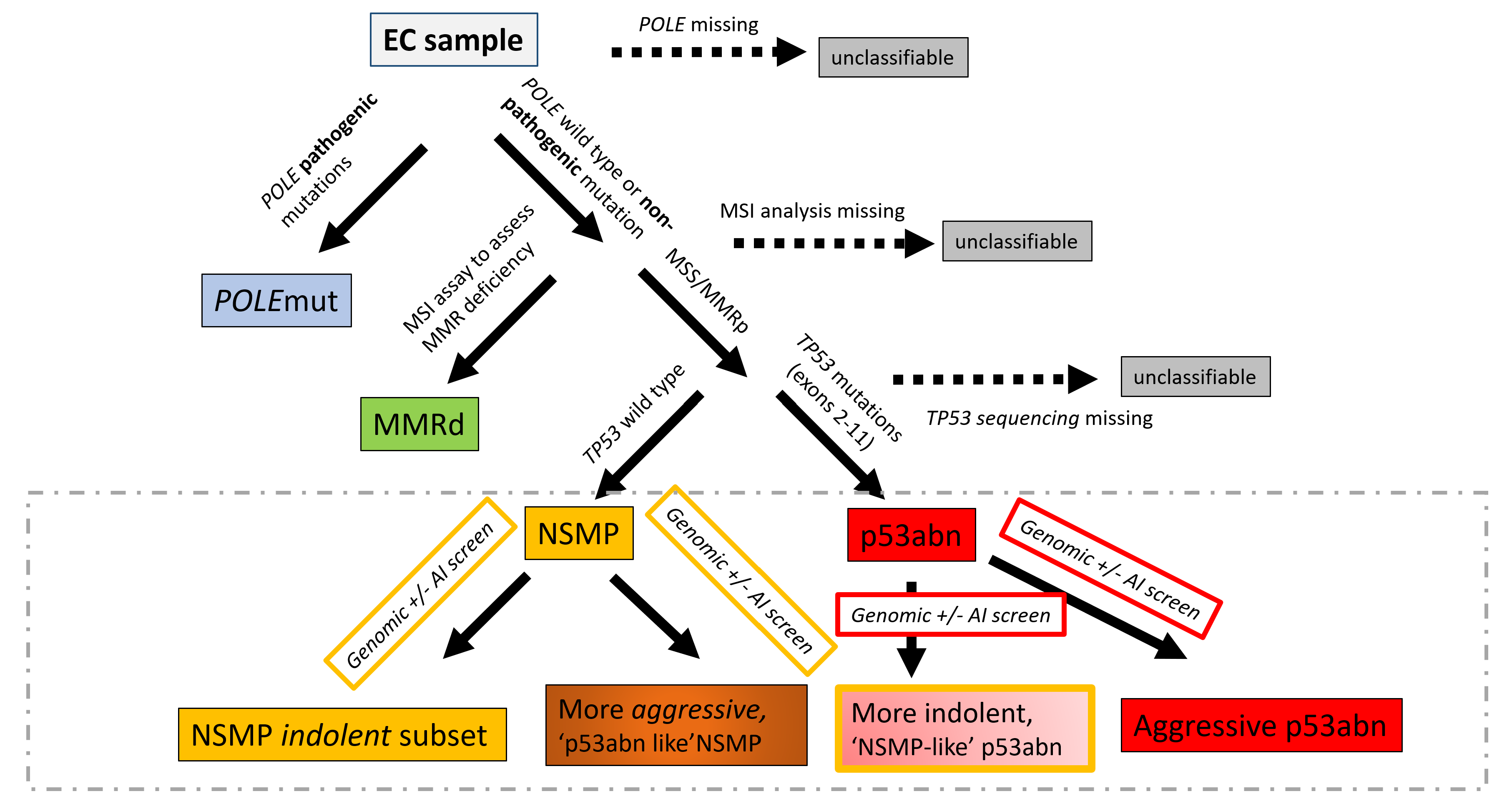
Endometrial cancer (EC) has four molecular subtypes with strong prognostic value and therapeutic implications. The most common subtype (NSMP; No Specific Molecular Profile) is assigned after exclusion of the defining features of the other three molecular subtypes and includes patients with heterogeneous clinical outcomes. In this study, we employ artificial intelligence (AI)-powered histopathology image analysis to differentiate between p53abn and NSMP EC subtypes and consequently identify a sub-group of NSMP EC patients that has markedly inferior progression-free and disease-specific survival (termed ‘p53abn-like NSMP’), in a discovery cohort of 368 patients and two independent validation cohorts of 290 and 614 from other centers. Shallow whole genome sequencing reveals a higher burden of copy number abnormalities in the ‘p53abn-like NSMP’ group compared to NSMP, suggesting that this group is biologically distinct compared to other NSMP ECs. Our work demonstrates the power of AI to detect prognostically different and otherwise unrecognizable subsets of EC where conventional and standard molecular or pathologic criteria fall short, refining image-based tumor classification. This study’s findings are applicable exclusively to females.
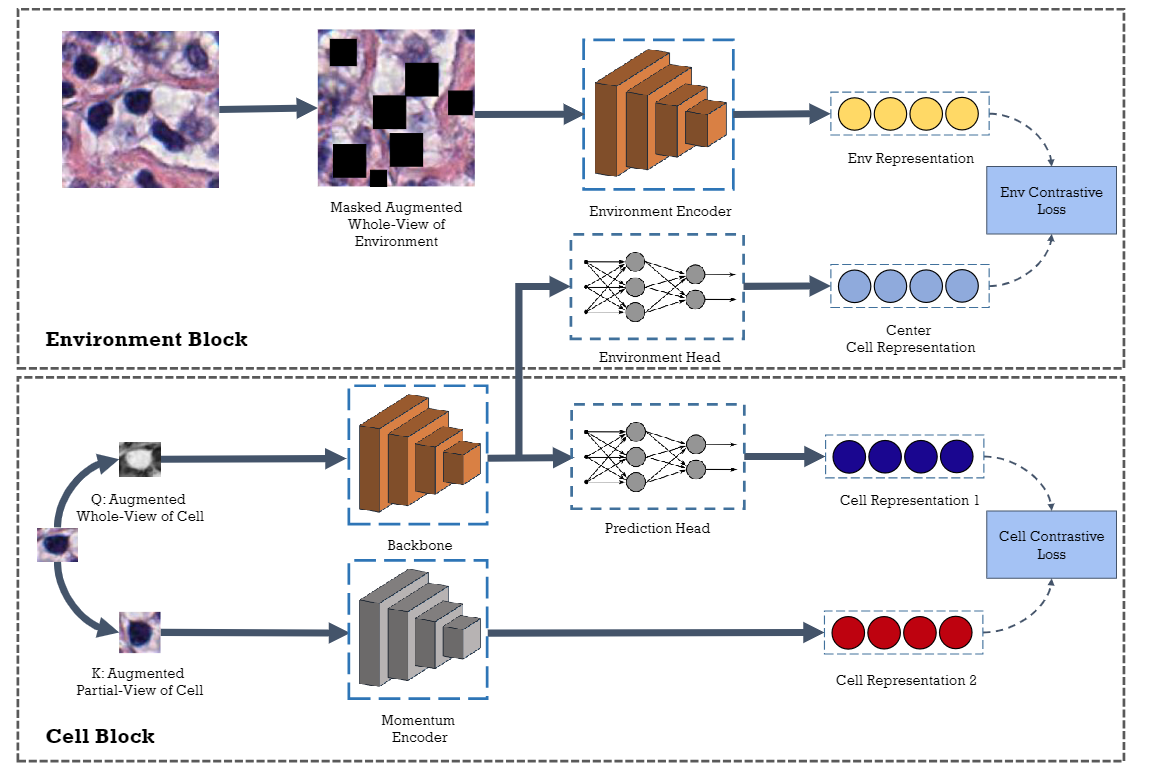
In clinical practice, many diagnosis tasks rely on the identification of cells in histopathology images. While supervised machine learning techniques require labels, providing manual cell annotations is time-consuming due to the large number of cells. In this paper, we propose a self-supervised framework (Volta) for cell representation learning in histopathology images using a novel technique that accounts for the cell's mutual relationship with its environment for improved cell representations. We subjected our model to extensive experiments on the data collected from multiple institutions around the world comprising of over 700,000 cells, four cancer types, and cell types ranging from three to six categories for each dataset. The results show that our model outperforms the state-of-the-art models in cell representation learning. To showcase the potential power of our proposed framework, we applied Volta to ovarian and endometrial cancers with very small sample sizes (10-20 samples) and demonstrated that our cell representations can be utilized to identify the known histotypes of ovarian cancer and provide novel insights that link histopathology and molecular subtypes of endometrial cancer. Unlike supervised deep learning models that require large sample sizes for training, we provide a framework that can empower new discoveries without any annotation data in situations where sample sizes are limited.
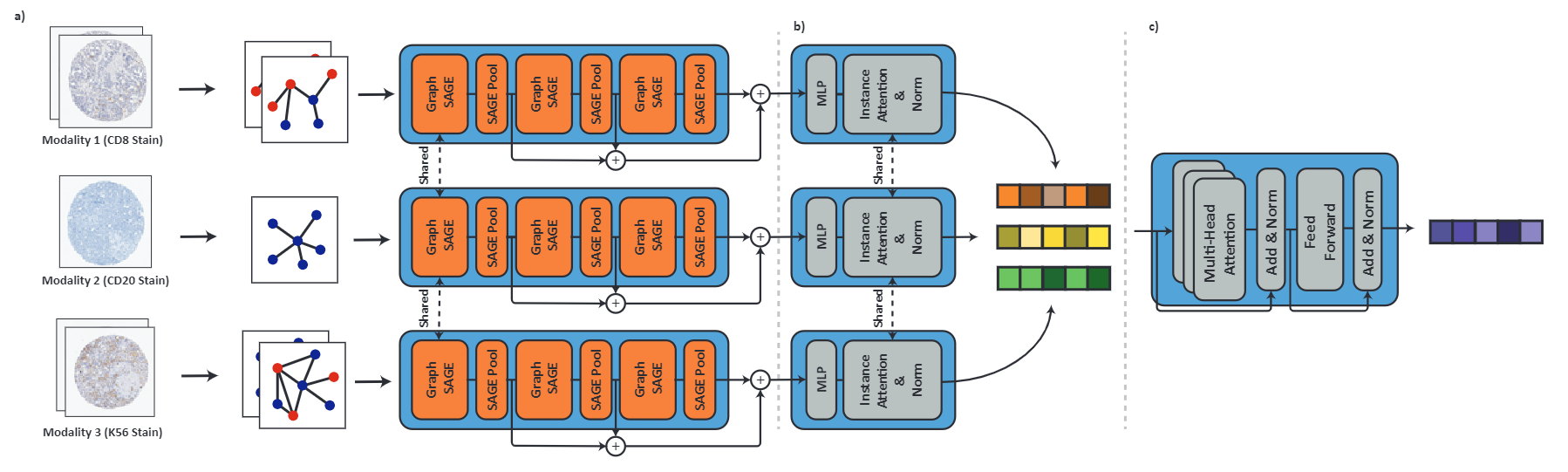
Processing giga-pixel whole slide histopathology images (WSI) is a computationally expensive task. Multiple instance learning (MIL) has become the conventional approach to process WSIs, in which these images are split into smaller patches for further processing. However, MIL-based techniques ignore explicit information about the individual cells within a patch. In this paper, by defining the novel concept of shared-context processing, we designed a multi-modal Graph Transformer (AMIGO) that uses the celluar graph within the tissue to provide a single representation for a patient while taking advantage of the hierarchical structure of the tissue, enabling a dynamic focus between cell-level and tissue-level information. We benchmarked the performance of our model against multiple state-of-the-art methods in survival prediction and showed that ours can significantly outperform all of them including hierarchical Vision Transformer (ViT). More importantly, we show that our model is strongly robust to missing information to an extent that it can achieve the same performance with as low as 20% of the data. Finally, in two different cancer datasets, we demonstrated that our model was able to stratify the patients into low-risk and high-risk groups while other state-of-the-art methods failed to achieve this goal. We also publish a large dataset of immunohistochemistry images (InUIT) containing 1,600 tissue microarray (TMA) cores from 188 patients along with their survival information, making it one of the largest publicly available datasets in this context.
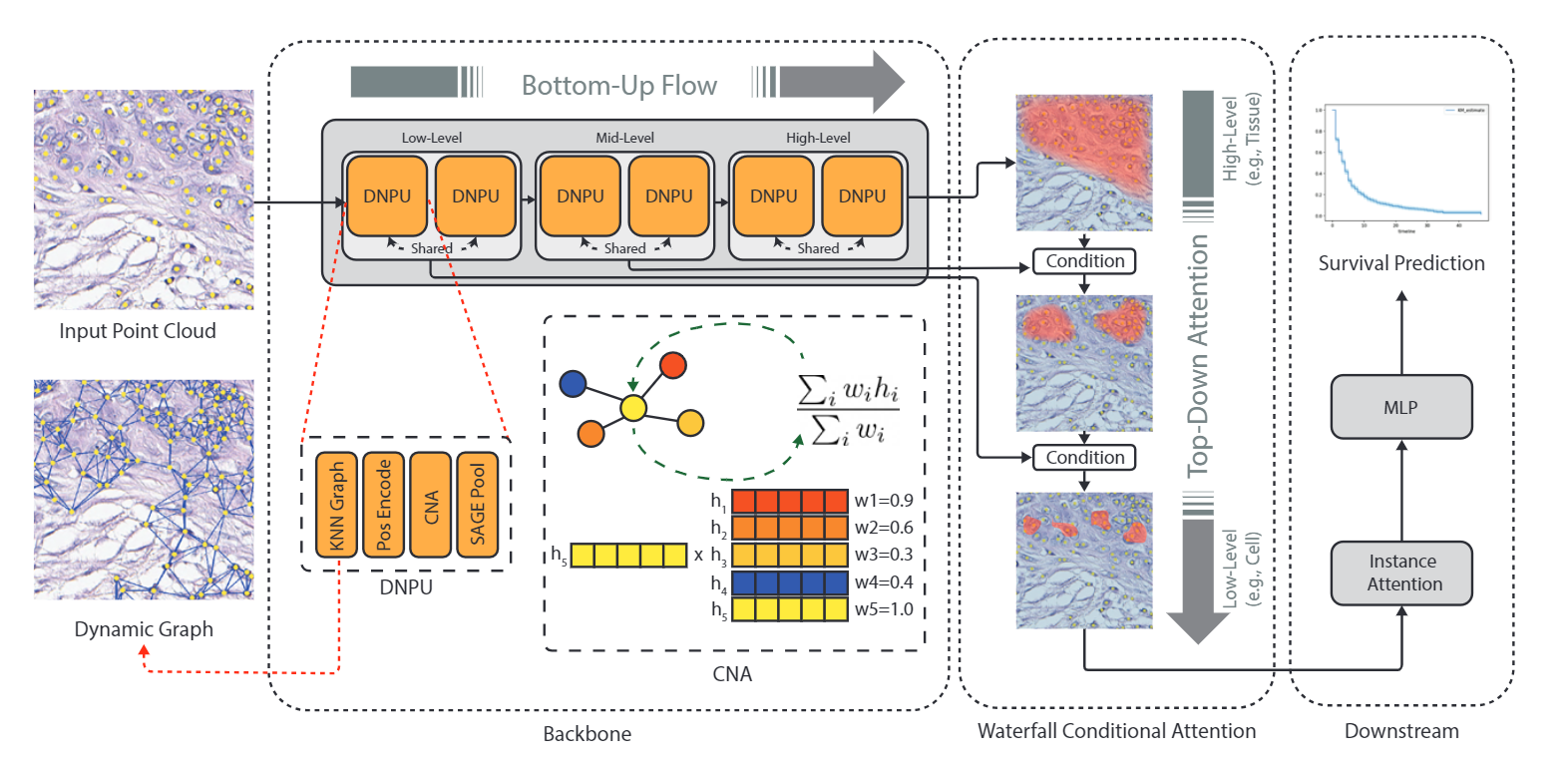
Predicting survival rates based on multi-gigapixel histopathology images is one of the most challenging tasks in digital pathology. Due to the computational complexities, Multiple Instance Learning (MIL) has become the conventional approach for this process as it breaks the image into smaller patches. However, this technique fails to account for the individual cells present in each patch, while they are the fundamental part of the tissue. In this work, we developed a novel dynamic and hierarchical point-cloud-based method Co-Pilot for the processing of cellular graphs extracted from routine histopathology images. By using bottom-up information propagation and top-down conditional attention, our model gains access to an adaptive focus across different levels of tissue hierarchy. Through comprehensive experiments, we demonstrate that our model can outperform all the state-of-the-art methods in survival prediction, including the hierarchical Vision Transformer (ViT), across three datasets and four metrics with only half of the parameters of the closest baseline. Importantly, our model is able to stratify the patients into different risk cohorts with statistically different outcomes across three large datasets, a task that was previously achievable only using genomic information. Furthermore, we publish a large dataset containing 873 cellular graphs from 188 patients, along with their survival information, making it one of the largest publicly available datasets in this context.
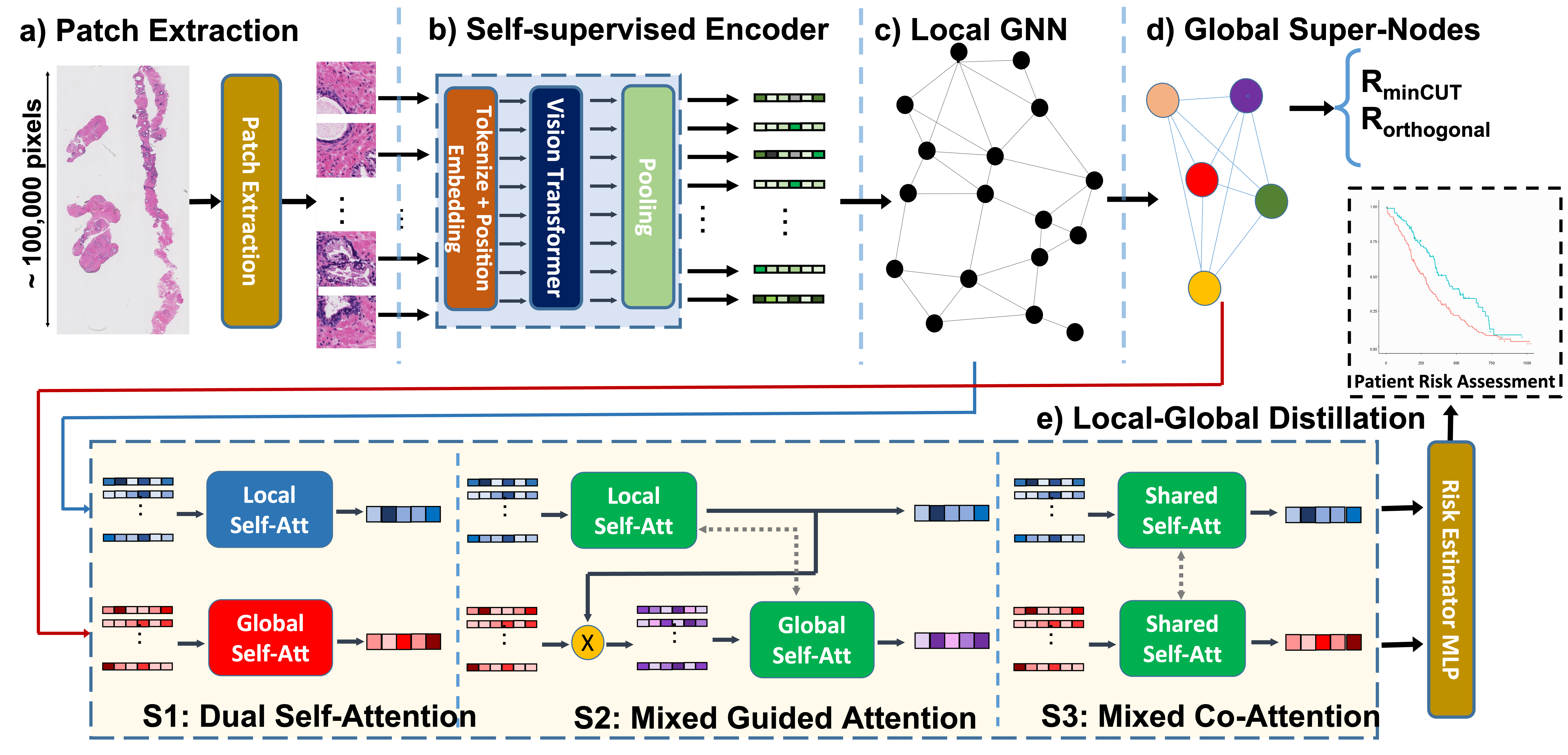
The utility of machine learning models in histopathology image analysis for disease diagnosis has been extensively studied. However, efforts to stratify patient risk are relatively under-explored. While most current techniques utilize small fields of view (so-called local features) to link histopathology images to patient outcome, in this work we investigate the combination of global (i.e., contextual) and local features in a graph-based neural network for patient risk stratification. The proposed network not only combines both fine and coarse histological patterns but also utilizes their interactions for improved risk stratification. Our results suggest that the proposed model is capable of stratifying patients into statistically significant risk groups (p < 0.01 across the two datasets) with actionable clinical utility.
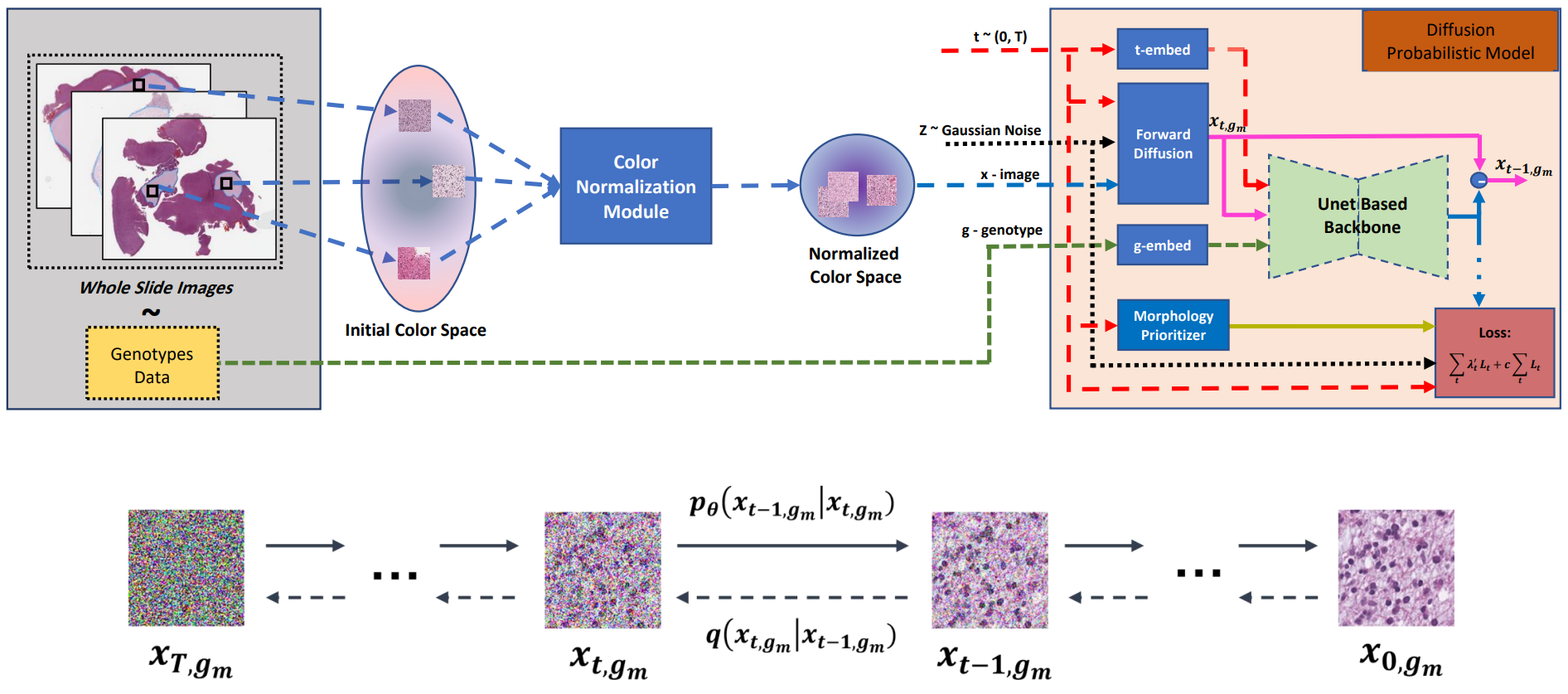
Visual microscopic study of diseased tissue by pathologists has been the cornerstone for cancer diagnosis and prognostication for more than a century. Recently, deep learning methods have made significant advances in the analysis and classification of tissue images. However, there has been limited work on the utility of such models in generating histopathology images. These synthetic images have several applications in pathology including utilities in education, proficiency testing, privacy, and data sharing. Recently, diffusion probabilistic models were introduced to generate high quality images. Here, for the first time, we investigate the potential use of such models along with prioritized morphology weighting and color normalization to synthesize high quality histopathology images of brain cancer. Our detailed results show that diffusion probabilistic models are capable of synthesizing a wide range of histopathology images and have superior performance compared to generative adversarial networks.
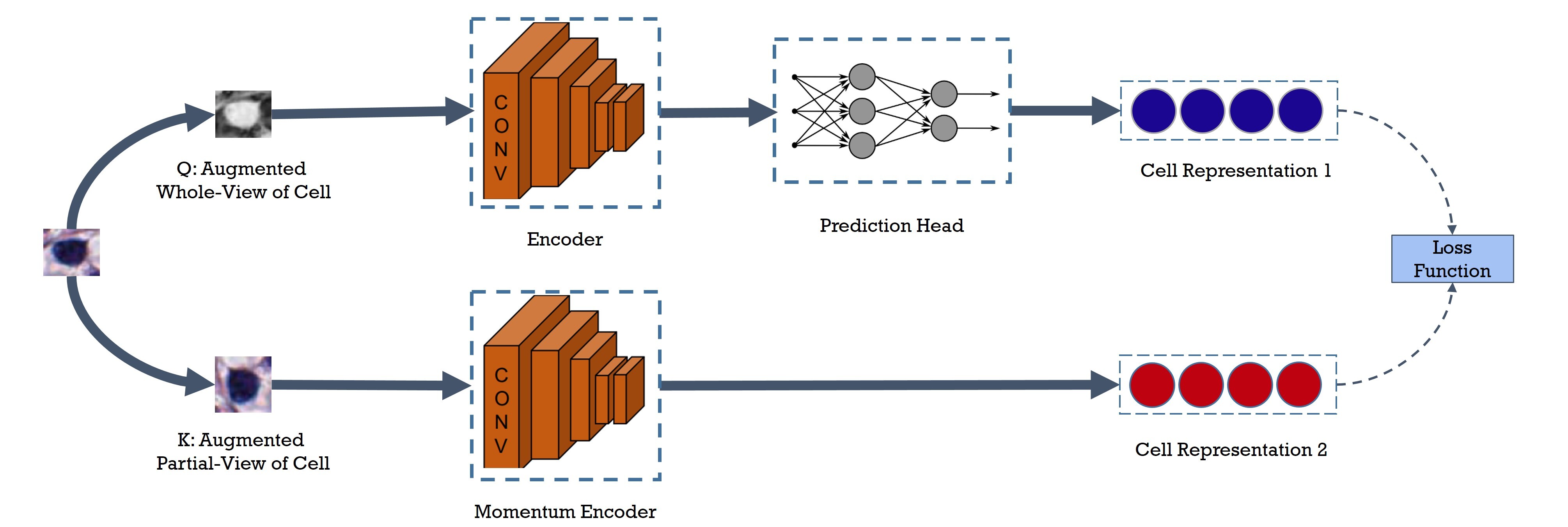
Cell identification within the H&E slides is an essential prerequisite that can pave the way towards further pathology analyses including tissue classification, cancer grading, and phenotype prediction. However, performing such a task using deep learning techniques requires a large cell-level annotated dataset. Although previous studies have investigated the performance of contrastive self-supervised methods in tissue classification, the utility of this class of algorithms in cell identification and clustering is still unknown. In this work, we investigated the utility of Self-Supervised Learning (SSL) in cell clustering by proposing the Contrastive Cell Representation Learning (CCRL) model. Through comprehensive comparisons, we show that this model can outperform all currently available cell clustering models by a large margin across two datasets from different tissue types. More interestingly, the results show that our proposed model worked well with a few number of cell categories while the utility of SSL models has been mainly shown in the context of natural image datasets with large numbers of classes (e.g., ImageNet). The unsupervised representation learning approach proposed in this research eliminates the time-consuming step of data annotation in cell classification tasks, which enables us to train our model on a much larger dataset compared to previous methods. Therefore, considering the promising outcome, this approach can open a new avenue to automatic cell representation learning.
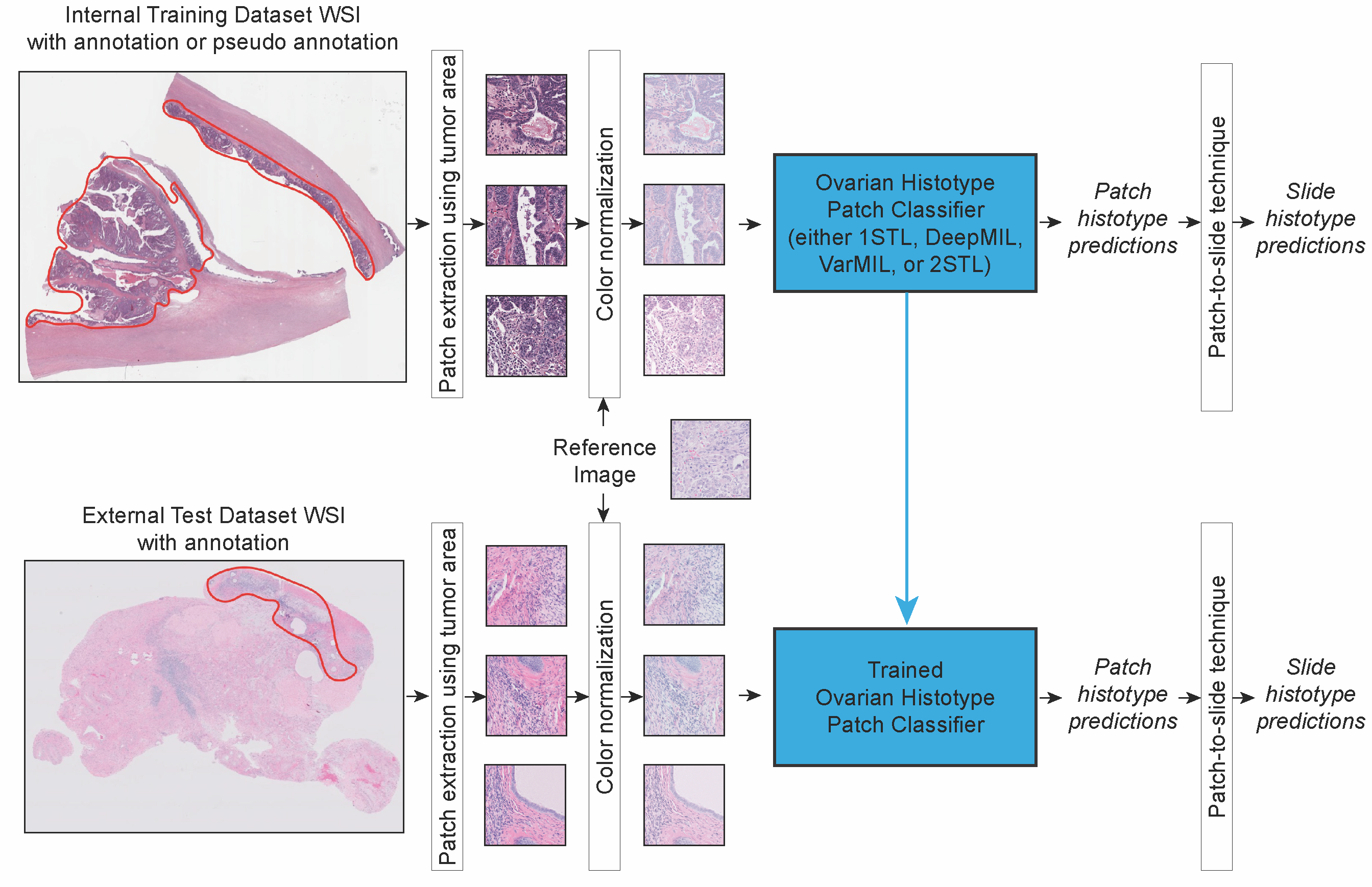
We compared four different deep convolutional neural networks for classifying H&E-stained images of epithelial ovarian carcinoma histotypes using the largest training dataset to date (948 slides corresponding to 485 patients), exploring techniques such as color normalization and partially balancing the histotypes. The best performing model, assessed on an independent test set of 60 patients from another institution, achieved a mean diagnostic concordance of 80.97% (Cohen’s kappa 0.7547). As well, in 4 of 8 cases misclassified by ML on the external testing dataset, two expert subspecialty pathologists rendered diagnoses, based on blind review of the WSIs, that agree with AI rather than the integrated reference diagnosis. Our results indicate that color normalization can reliably improve AI-based diagnosis of WSIs sourced from multiple centers, and specifically that an ML-based ovarian carcinoma classifier is ready for clinical validation studies as an adjunct for informing histotype diagnosis.
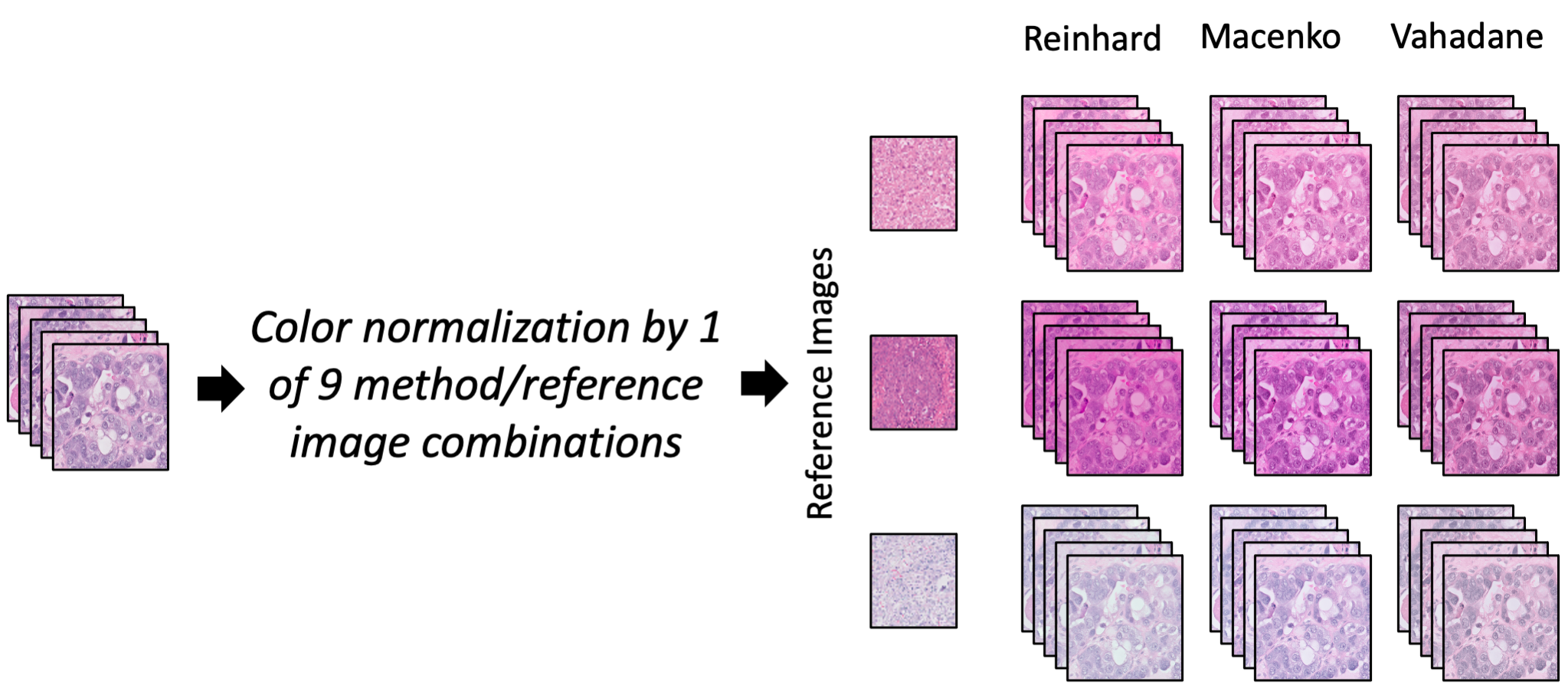
The color variation of hematoxylin and eosin (H&E)-stained tissues has presented a challenge for applications of artificial intelligence (AI) in digital pathology. In this study, we investigated eight color normalization algorithms for AI-based classification of H&E-stained histopathology slides, in the context of both using images from one center and from multiple centers. Our results show that color normalization does not consistently improve classification performance when both training and testing data are from a single center. However, using four multi-center datasets of two cancer types, we show that color normalization can significantly improve the classification of images from external datasets (ovarian cancer 0.25 AUC increase, p=1.6e-05, pleural cancer 0.21 AUC increase, p=1.4e-10). Furthermore, we introduce a novel augmentation strategy by mixing color-normalized images using three easily accessible algorithms that consistently improves the diagnosis of test images from external centers, even when the individual normalization methods had varied results.
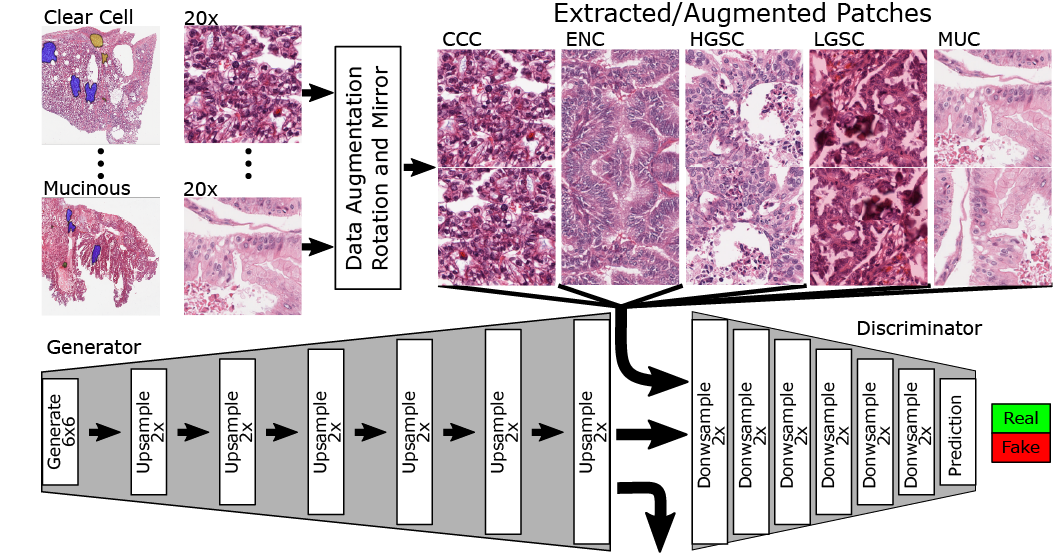
The purpose of this project is to showcase the capacity of generative adversarial networks (GAN) in synthesizing realistic looking histopathology images. Our findings have important applications in proficiency testing of medical practitioners and quality assurance in clinical laboratories. Furthermore, training of computer-aided diagnostic systems can benefit from synthetic images where labeled datasets are limited (e.g., rare cancers). We have created a publicly available demo website where clinicians and researchers can attempt questions from the image survey.
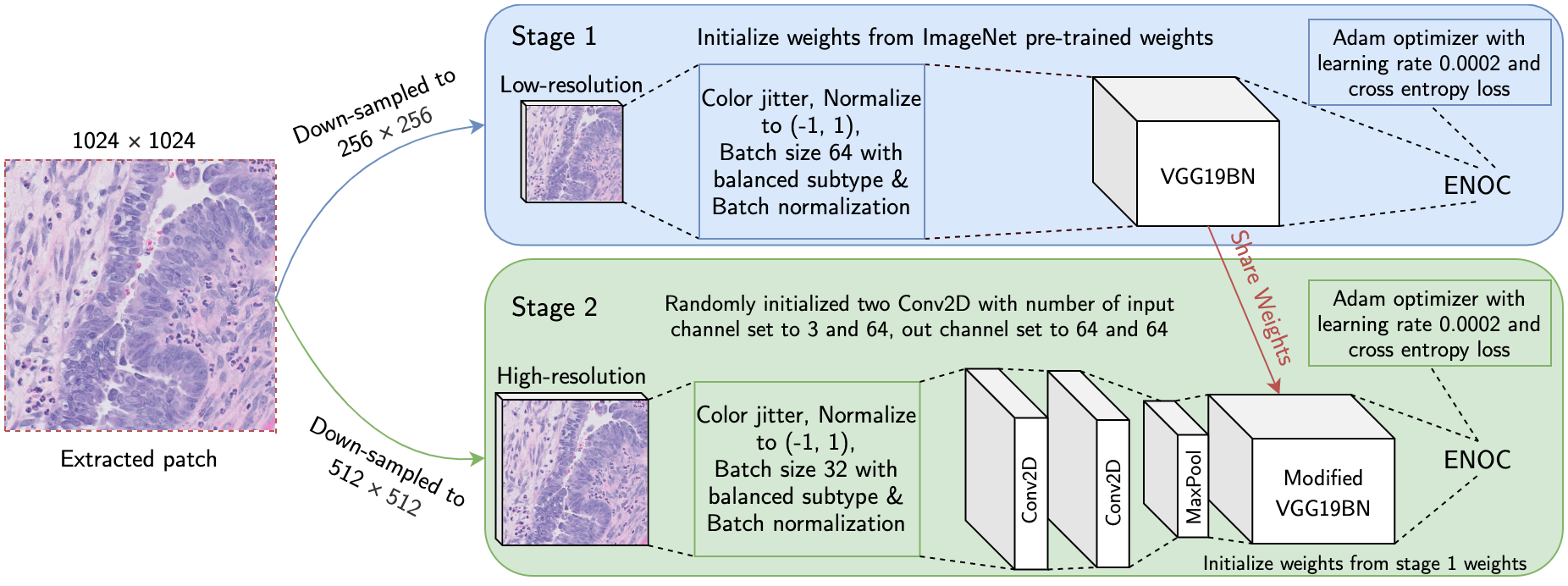
Ovarian cancer is the most lethal cancer of the female reproductive organs. There are 5 major histological subtypes of epithelial ovarian cancer, each with distinct morphological, genetic, and clinical features. Currently, these histotypes are determined by a pathologist's microscopic examination of tumor whole-slide images (WSI). This process has been hampered by poor inter-observer agreement (Cohen’s kappa 0.54-0.67). We utilized a two-stage deep transfer learning algorithm based on convolutional neural networks (CNN) and progressive resizing for automatic classification of epithelial ovarian carcinoma WSIs. The proposed algorithm achieved a mean accuracy of 87.54% and Cohen's kappa of 0.8106 in the slide-level classification of 305 WSIs; performing better than a standard CNN and pathologists without gynecology-specific training.
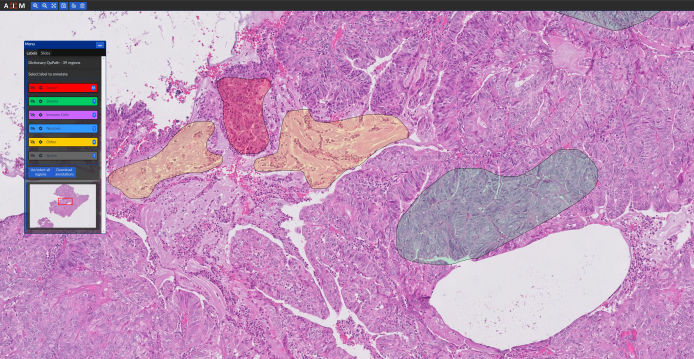
In order to build and evaluate machine learning (ML) models, histopathology slide images need to be annotated by pathologists. Furthermore, the evaluation of ML models needs a conveninet platform for viweing the results. Our group have developed a web-based application (named “cPathPortal”) for quick and intuitive annotation of histopathology images that works with tablets with stylus. cPathPortal is built on top of OMERO (https://www.openmicroscopy.org/omero/) and allows the users to load histopathology images, zoom into different sections and annotate them. Our intention is to release cPathPortal as an open-source platform in the near future. Please contact us if you would like to deploy and test cPathPortal on your own servers.
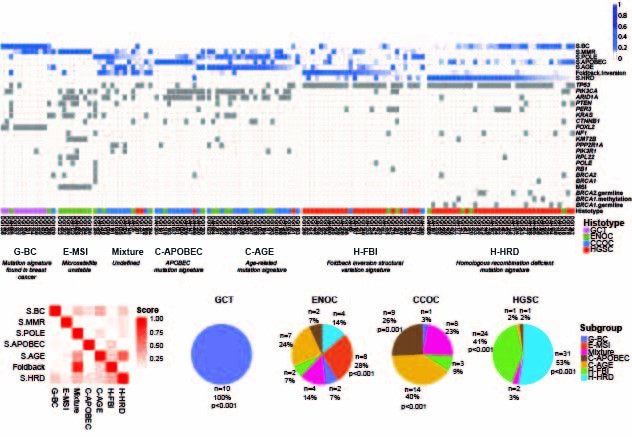
we stratified ovarian cancer histotypes into novel genomic subtypes. This major discovery was built upon novel approach of integrating computation of point mutation and structural variation signatures that provided a potent, genome-based discriminant biomarker across the spectra of ovarian cancer histotypes. The most striking finding was a previously unreported poor outcome group representing ~50% of high grade serous ovarian cancer (HGSOC) cases. The poor outcome group was characterized by specific types of genomic structural rearrangement reflective of micro homology mediated end joining (MMEJ) as an active double strand break repair process that was notably absent from the other HGSOC cases.

We conducted an integrative genomic analysis on a large cohort of patients with de novo DLBCL, drawn from a population registry, that were treated uniformly with modern immune-chemotherapy (R-CHOP). This study provides unprecedented insight into mutation-associated changes in membrane physiology linked to cancer. We identified and characterized TMEM30A as a novel tumor suppressor gene whose loss is exclusively found in DLBCL and associated with favourable outcome after immuno-chemotherapy.

Kronos is a software platform for facilitating the development and execution of modular, auditable, and distributable bioinformatics workflows. Kronos obviates the need for explicit coding of workflows by compiling a text configuration file into executable Python applications. The Kronos platform provides a standard framework for developers to implement custom tools, reuse existing tools, and contribute to the community at large.
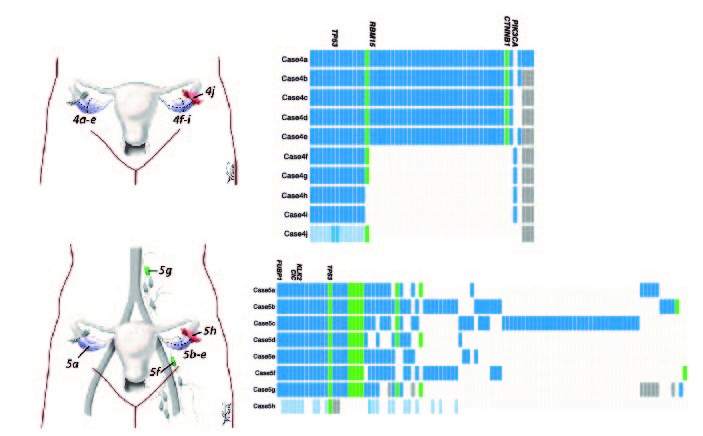
The first world study on quantifying the degree of clonal diversity and evolution in primary untreated high-grade serous ovarian cancers (HGSOC). In this study, we measured the degree of genomic diversity within primary, untreated HGSCs to examine the natural state of tumour evolution prior to therapy. Our results demonstrate extensive intra-tumoral mutational, copy number and expression heterogeneity in HGSCs, thus illuminating a challenge for application of sequencing tumor genomes in the context of personalized, precision medicine. Although HGSCs are considered a single disease, they exhibit individual evolutionary trajectories that will require consideration in future therapeutic solutions.
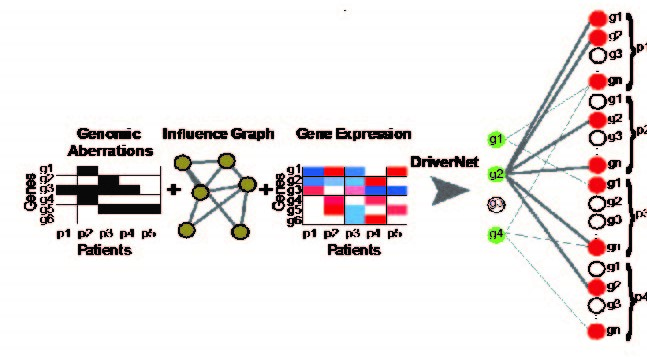
We introduce a novel computational framework, DriverNet, to identify likely driver mutations by virtue of their effect on mRNA expression networks. Application to four cancer datasets reveals the prevalence of rare candidate driver mutations associated with disrupted transcriptional networks and a simultaneous modulation of oncogenic and metabolic networks, induced by copy number co-modification of adjacent oncogenic and metabolic drivers.

